mkdos.py (DOS)¶
mkdos is a python module to work with DOSCAR files. It consists of a class “dos” that lets you read and output information. Most important information is stored in self.ens, self.tot, and self.pdos. Information is retrievable using the get_val() methods.
Python usage¶
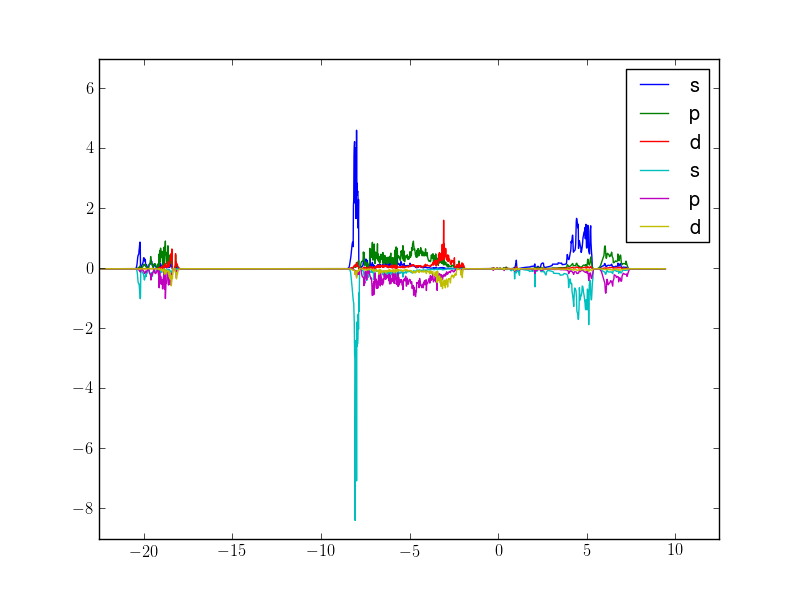
Here is an example script to display s,p,d PDOS summed over atoms 1-4, separating spin-up and spin-down:
#!/usr/bin/env python
from mkdos import dos
import matplotlib.pyplot as plt
dd=dos('DOSCAR')
arr,lab=dd.get_perl(range(4))
colors='b g r c m y k'.split()
for ispin,spin in enumerate([1,-1]):
for l in range(3):
plt.plot(dd.ens,spin*arr[ispin,l,:],colors[ispin*3+l])
ax=plt.gca()
ax.legend(lab[1]*2)
plt.show()
Which outputs this:
- Short methods descriptions (see docstrings for details):
init Input a DOSCAR, optionally specify spin-polarization, and whether to shift fermi energy to zero
get_energies returns vector of energies
get_peratom returns array of PDOS for each atom. You can specify to sum spin up and spin dn.
get_perl returns array of PDOS for each orbital quantum number. You can specify which atoms to sum over, and/or to sum spin-up and spin-dn.
get_perm returns array of PDOS for each l_m for a given l. You can specify which atoms and/or summing spin-up and spin-dn.
get_total returns vector of total DOS
get_totspin returns vector of total DOS spin-up then spin-dn
set_pdos shouldn’t be necessary to use; it’s just for reading the DOSCAR
set_total shouldn’t be necessary to use; it’s just for reading the DOSCAR
shift_fermi shouldn’t be necessary to use after instantiation.
- Notes:
get_PDOS() methods return a tuple of the form (out,label), where label tells you what it is you got
spin-polarized get_PDOS()[0] may return a 3d array, where the first index is for spin up or down.
Note that when identifying atoms, indexing is from 0. This is a very easy bug to squish.
If you want to plot s,p,d of atoms 1,2, it is easiest to do something like:
np.concatenate((dd.get_perl(0,True)[0],dd.get_perl(1,True)[0]),axis=0)
Command-line usage¶
Like boomerang and periodica, there is a command-line interface, which is used via command-line arguments. It is easily piped to gracey.
filename can be inserted anywhere in the arguments. Default is DOSCAR, then DOSCAR.static.
noshiftfermi: Do not shift energies to set fermi energy to zero
sp: Try to read DOSCAR with spin polarization off (sp=1) or on (sp=2); default reads INCAR to find out.
- Only one of the following; they are gone through in order:
total: Print total DOS (each line: en, totdos)
atom: Print per-atom DOS (each line: en, at1, at2, …)
l: Print per-l DOS (each line: en, s, p, d). Note that spin-up and spin-down are added, and there is no option on the command-line to choose only some atoms. Python has that capability.
m=int: Print per-m DOS for the given l=int (0,1,2=s,p,d). Output is each line: en,m1,m2,m3…. Note that spin-up and spin-down are added, and there is no option on the command-line to choose only some atoms. Python has that capability. Also the order of m is not what you may expect, but is the same as in DOSCAR. See http://cms.mpi.univie.ac.at/vasp-forum/forum_viewtopic.php?3.306
Here is an example:
mkdos.py DOSCAR -total | gracey.py