VASP SrVO3 Density of states¶
Here we compute the density-of-states using VASP.
Total Density-of-states¶
INPUT: INCAR | KPOINTS | POTCAR | POSCAR
OUTPUT: CONTCAR | OSZICAR | OUTCAR | DOSCAR | PROCAR | vasp.out
This calculation needs to be a static run (ie. no structural relaxation NSW=0). Please see relaxation introduction for a basic description of the four required files. A typical INCAR would be as follows:
ISPIN = 2 # Spin polarized calculation
MAGMOM= 0 4 0 0 0 # Initialization of moments
PREC = med # Sets cutoffs and grids
NSW=0 # Don't move the atoms
ISMEAR = -5 # Use the tetrahedron method
ENCUT=375 # Plane wave cuttoff
NEDOS=5000 # Number of energy points for DOS
This would produce a file called DOSCAR which has the total density of states and the projected DOS if requested. The format of the DOSCAR is as follows:
5 5 0 0 # first five lines not terribly useful...
0.1148420E+02 0.3857953E-09 0.3857953E-09 0.3857953E-09 0.5000000E-15
1.000000000000000E-004
CAR
unknown system
12.00000000 -3.00000000 5000 4.96643483 1.00000000 # Total_elec unknown NEDOS Efermi unknown
-3.000 0.0000E+00 0.0000E+00 0.7000E+01 0.7000E+01 # Energy tdos_up tdos_dn cdos_up cdos_dn
-2.997 0.0000E+00 0.0000E+00 0.7000E+01 0.7000E+01 # ...
-2.994 0.0000E+00 0.0000E+00 0.7000E+01 0.7000E+01 # ...
...
One problem you will have is that VASP defaults to making the mim/max energy of the DOS as the entire range of the spectrum which you rarely care about. If you use many points (ie. NEDOS) you will still resolve things at the expense of huge files. The best answer is to finish writing our own software to compute the DOS as a postprocessing step. In the meantime, one can just keep the WAVECAR and quickly rerun to recompute the DOS in a smaller energy range. Do a quick plot of the DOS to find out what max/min energy you want. Be careful that the plotting code does not shift the results by the Fermi energy. You can extract the total DOS from the DOSCAR file using a very antiquated script called dformat.py and then plot it however you like. dformat.py desperately needs to be rewritten:
$ dformat.py total noshiftfermi > dos.out
Now you can add these two lines to the INCAR and rerun:
EMIN=-3 # Minimum energy for computing DOS
EMAX=12 # Maximum energy for computing DOS
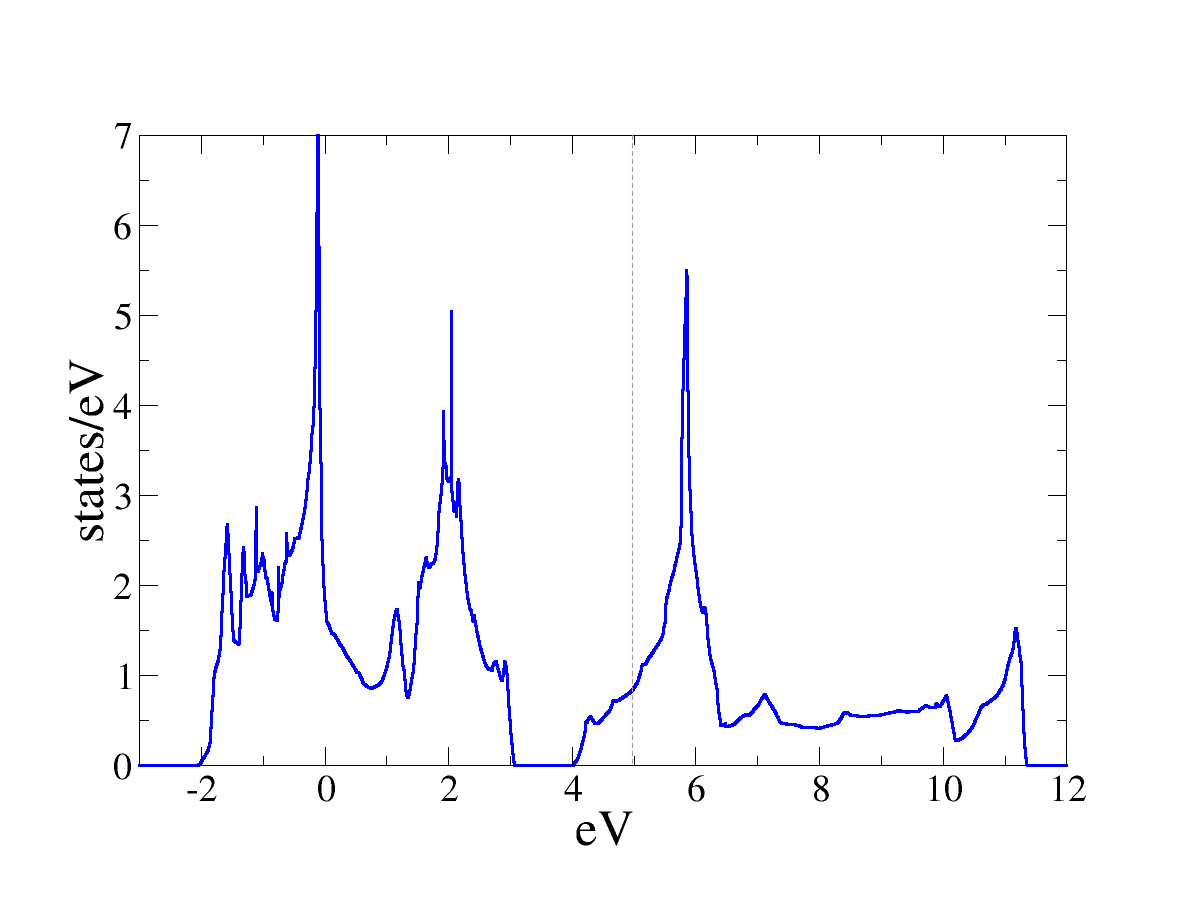
DOS. This is the density of states for SrVO3. I only plotted one
spin component as the calculaion found a non-spin-polarized solution.
EPS
Plotting Script¶
Partial Density-of-states¶
If you want to have the projected DOS, you need to set the LORBIT parameter in the INCAR. You should probably be in the habbit of turning this on.
LORBIT=11 # Compute projections along with phase factors
This will result in the creation of the PROCAR file which has the projections onto local orbitals for each k-point and each band. We will not worry about the PROCAR at this point.