Linear Response U¶
Introduction¶
Here I (Eric) discuss computing Hubbard U via the linear response approach in VASP. You should first read M. Cococcioni and S. de Gironcoli, Phys. Rev. B 71, 035105 (2005) to understand the theory. The basic idea is that we are applying a potential to the correlated (I’ll refer to them as d states in this document) levels of a single site and observing how this changes the d occupancies on all the sites. This tells us about the screened on-site Coulomb interaction.
- In principal we need to perform three types of calculations:
normal DFT calculation (
 )
)charge-self-consistent calculations for a series of finite
 (“interacting” response)
(“interacting” response)non-charge-self-consistent calculations for a series of finite
(“bare” response)
The first type gives us the number of d electrons ( ) and
the charge density for the unperturbed () system. For
the second and third types we read in this charge density via the
CHGCAR file (which also includes the density matrices) and apply a
finite . In the second type we allow the charge density
to relax to help screen this perturbation, while for the third type we
do not.
) and
the charge density for the unperturbed () system. For
the second and third types we read in this charge density via the
CHGCAR file (which also includes the density matrices) and apply a
finite . In the second type we allow the charge density
to relax to help screen this perturbation, while for the third type we
do not.
I typically use values of  0.08,
0.05, and 0.02, which in my experience usually falls
within the linear regime of vs. .
0.08,
0.05, and 0.02, which in my experience usually falls
within the linear regime of vs. .
1-shot versus 2-shot approaches¶
Note that in the Cococcioni paper (and in Quantum ESPRESSO) people
typically look after the first electronic iteraction (i.e., before the
charge density is updated for the first time) to obtain the bare
response. We call this the “1-shot” approach. In principle this is a
fine thing to do provided one uses exact diagonalization or
sufficiently converges the iterative diagonalization before updating
the charge density for the first time (e.g. by making NELMDL more
negative). However, empirically we find that this approach DOES
NOT work in VASP and underestimates U by approximately 1 eV. After
extensive testing, our best understanding is that VASP is doing
something strange for the charge-self-consistent run that is enabling
more screening of than should be present after the
first electronic step if the charge density were really not yet
changed. Therefore, one should do separate calculations to obtain the
bare and interacting responses, which we call the “2-shot” approach.
What VASP executable should I use?¶
vasp.5.3_wei¶
We have a modified version of VASP (vasp.5.3_wei) written by Wei Xie
in Dane Morgan’s group that implements the potential perturbation
. One can successfully compute U using this code, but
there is some extra work involved. For bare
(non-charge-self-consistent) runs the density matrix printed out by
VASP is just that read in from the CHGCAR file initially and does not
reflect the effects of the finite . If we were just
doing something like computing a DFT+U band structure, this would be a
reasonable thing for VASP to do since we know the potential based on
that density and density matrices is already correct. However, in our
case this is problematic since is actually changing the
density matrices in some way about which we care.
Therefore, one needs to play a trick of saving the WAVECAR from the bare run, reading it into a 1-electronic-step (NELM=1) charge-self-consistent run to compute and print the actual density matrices corresponding to the WAVECAR. This takes some extra time and requires more (temporary) hard disk storage, so it is undesirable. However, the approach does work so let me know if you want to use it and I can explain how to use the executable.
LDAUTYPE=3 in VASP 5¶
We discovered computing linear response U with the 2-shot approach is
actually a hidden feature in VASP 5 using the LDAUTYPE=3 tag. The only
information on this is on this VASP forum post. This
post includes instructions on how to run the calculations. Basically
you need to set LDAUTYPE=3 as well as the LDAUU and LDAUJ parameters,
which are now used as the parameters for the up and
down spin channels, respectively, instead of the U and J values. For
computing U one should apply the potential to both spin channels so
the parameters LDAUU and LDAUJ should be identical.
One subtlety is that for this method the site at which the perturbation is applied must be listed as its own atomic species. So the POSCAR and POTCAR need to be modified to reflect this. For example, for iron (Fe) if we are using an 8-atom unit cell we should split this up into 1 species with 1 ion and 1 species with 7 ions and include the Fe pseudopotential twice in POTCAR. LDAUU and LDAUJ should have finite value only for this first atomic species since we only want to apply the perturbation to a single site as opposed to all of them.
Another very important subtetly we have found empirically is a
difference in sign convention for the LDAUTYPE=3 method. Basically we
believe the applied is actually  , so you
should flip the signs of all your . This will restore
the expected behavior that negative should lead to
increased on this site of the perturbation.
, so you
should flip the signs of all your . This will restore
the expected behavior that negative should lead to
increased on this site of the perturbation.
For LDAUTYPE=3 calculations the density matrices themselves are not printed but one can just find the trace (which is all we need) from bottom of the OUTCAR file (making sure LORBIT=11 so this is computed).
Calculating U¶
One can perform linear regression on the
vs. data to get the elements of the first row of the
response matrices  (when using data from the interacting
runs) and
(when using data from the interacting
runs) and  (when using data from the bare runs). To fill
in the rest of the upper right half of each matrix, one can use the
symmetry of the lattice since each site in the supercell is related to
the perturbed site via a translation vector of the primitive unit
cell. Finally, one can fill in the bottom left half of the matrices
trivially since these matrices are symmetrics again due to the
symmetry of the lattice.
(when using data from the bare runs). To fill
in the rest of the upper right half of each matrix, one can use the
symmetry of the lattice since each site in the supercell is related to
the perturbed site via a translation vector of the primitive unit
cell. Finally, one can fill in the bottom left half of the matrices
trivially since these matrices are symmetrics again due to the
symmetry of the lattice.
At the end of the day, one looks at the first diagonal entry of
 to get U (in the same units as
, which are eV).
to get U (in the same units as
, which are eV).
Important: One needs to keep increasing the size of the supercell for these calculations until the value of U stops changing.
resp_mat.f90¶
There is a code called resp_mat.f90 written by Cococcioni that you can find by searching around Quantum ESPRESSO tutorials for LDA+U. This code will do the postprocessing computation described in the previous section. It also has implemented the option to explicit enforce charge neutrality (see his paper) to speed up convergence with respect to supercell. In addition, it has implemented the extrapolation to larger supercell also discussed in that paper. I have found this code to be useful to check my own results. Documentation is in the comments of the source code, but let me know if you have any questions. One word of caution: at some point I think I saw a post in the Quantum ESPRESSO forum suggesting there might be a bug in this code for hexagonal unit cells.
Some example data¶
Here is some example data for ferromagnetic Fe for a 2x2x2 supercell of the primitive bcc unit cell using the 2-shot approach.
Note: For this case I am not enforcing charge neutrality or extrapolating to larger unit cell.
INCAR:
LWAVE = .FALSE.
LCHARG = .TRUE.
NELM = 50
ISYM = 0
ISPIN = 2
LMAXMIX = 4
LASPH = .TRUE.
LORBIT = 11
EDIFF = 1E-6
ISMEAR = -5
PREC = Accurate
ENCUT = 500.000
NEDOS = 2000
MAGMOM = 1*2 7*2
LDAU = .TRUE.
LDAUTYPE = 3
LDAUPRINT = 2
LDAUU = 0.0 0
LDAUJ = 0.0 0
LDAUL = 2 -1
 bare INCAR:
bare INCAR:
ICHARG = 11
LWAVE = .FALSE.
LCHARG = .FALSE.
NELM = 50
ISYM = 0
ISPIN = 2
LMAXMIX = 4
LASPH = .TRUE.
LORBIT = 11
EDIFF = 1E-6
ISMEAR = -5
PREC = Accurate
ENCUT = 500.000
NEDOS = 2000
MAGMOM = 1*2 7*2
LDAU = .TRUE.
LDAUTYPE = 3
LDAUPRINT = 2
LDAUU = -0.05 0
LDAUJ = -0.05 0
LDAUL = 2 -1
interacting INCAR:
ISTART = 0
ICHARG = 1
LWAVE = .FALSE.
LCHARG = .FALSE.
NELM = 50
ISYM = 0
ISPIN = 2
LMAXMIX = 4
LASPH = .TRUE.
LORBIT = 11
EDIFF = 1E-6
ISMEAR = -5
PREC = Accurate
ENCUT = 500.000
NEDOS = 2000
MAGMOM = 1*2 7*2
LDAU = .TRUE.
LDAUTYPE = 3
LDAUPRINT = 2
LDAUU = -0.05 0
LDAUJ = -0.05 0
LDAUL = 2 -1
Note that LDAUU and LDAUJ use -0.05 instead of 0.05 due to the different sign convention mentioned above.
Here I plot vs for each of the 8 Fe ions in
the supercell. Solid (dashed) lines correspond to the bare
(interacting) response. The line colors correspond to different Fe ion
indices.

First of all you can see that the magnitude of the changes of
is much larger for the bare case than the interacting
case, which makes sense since for the former the charge density is
unable to screen the perturbation. Secondly, you can notice that for
the on-site response (that of ion 1, the site at which
is applied, in blue) increases for negative
, which makes sense since lowering the potential
encourages more electrons to occupy the orbitals on that
site. Finally, you can observe that the response for both the bare and
the interacting cases are largest for ion 1 and decay away from this
site. You might notice that in this system the interacting response is
very small for all the ions other than the first, which might be
explained by the fact that we have a metallic system so the screening
is very efficient.
-1.2075 |
0.1554 |
0.1430 |
0.1312 |
0.1419 |
0.1253 |
0.1403 |
0.1581 |
0.1554 |
-1.2075 |
0.1312 |
0.1430 |
0.1253 |
0.1419 |
0.1581 |
0.1403 |
0.1430 |
0.1312 |
-1.2075 |
0.1554 |
0.1403 |
0.1581 |
0.1419 |
0.1253 |
0.1312 |
0.1430 |
0.1554 |
-1.2075 |
0.1581 |
0.1403 |
0.1253 |
0.1419 |
0.1419 |
0.1253 |
0.1403 |
0.1581 |
-1.2075 |
0.1554 |
0.1430 |
0.1312 |
0.1253 |
0.1419 |
0.1581 |
0.1403 |
0.1554 |
-1.2075 |
0.1312 |
0.1430 |
0.1403 |
0.1581 |
0.1419 |
0.1253 |
0.1430 |
0.1312 |
-1.2075 |
0.1554 |
0.1581 |
0.1403 |
0.1253 |
0.1419 |
0.1312 |
0.1430 |
0.1554 |
-1.2075 |
-0.2054 |
0.0075 |
0.0032 |
0.0108 |
0.0086 |
0.0048 |
0.0070 |
0.0113 |
0.0075 |
-0.2054 |
0.0108 |
0.0032 |
0.0048 |
0.0086 |
0.0113 |
0.0070 |
0.0032 |
0.0108 |
-0.2054 |
0.0075 |
0.0070 |
0.0113 |
0.0086 |
0.0048 |
0.0108 |
0.0032 |
0.0075 |
-0.2054 |
0.0113 |
0.0070 |
0.0048 |
0.0086 |
0.0086 |
0.0048 |
0.0070 |
0.0113 |
-0.2054 |
0.0075 |
0.0032 |
0.0108 |
0.0048 |
0.0086 |
0.0113 |
0.0070 |
0.0075 |
-0.2054 |
0.0108 |
0.0032 |
0.0070 |
0.0113 |
0.0086 |
0.0048 |
0.0032 |
0.0108 |
-0.2054 |
0.0075 |
0.0113 |
0.0070 |
0.0048 |
0.0086 |
0.0108 |
0.0032 |
0.0075 |
-0.2054 |
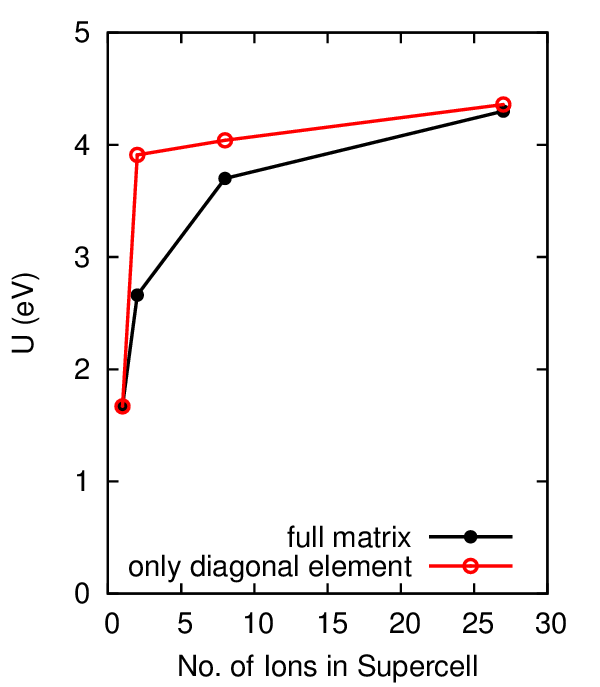
Here is a plot of the computed U in eV versus the size of the
supercell. You can see that U increases with supercell size and starts
to saturate a bit above 4 eV. The results from the full matrix
inversion are shown in black, while the results one obtains from only
using the diagonal elements of and are
shown in red.